
Dec 15, 2025
Sterility Assays in Drug and Device Manufacturing
Sterility and endotoxin testing define release safety in biologics, ensuring microbial control, regulatory compliance, and patient protection.
 \
\
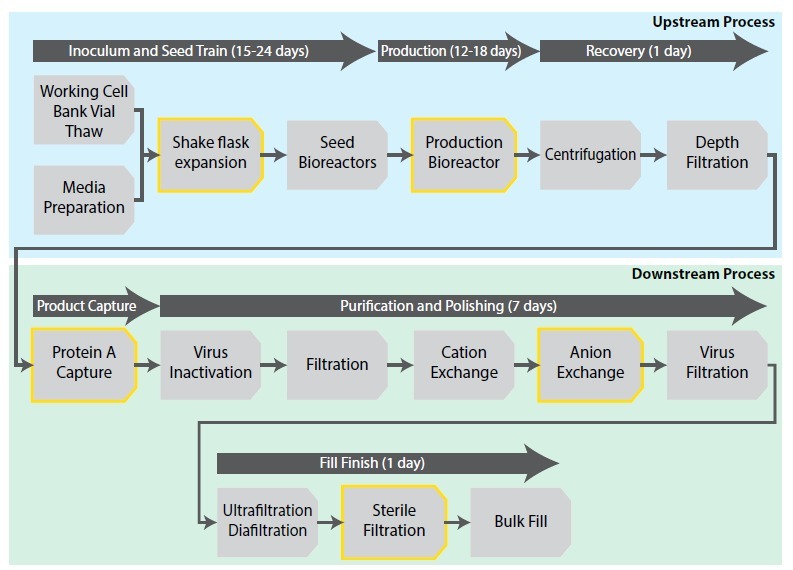
Figure 1. Process flow diagram of a monoclonal Antibody biomanufacturing process. Yellow highlights denote the checkpoints for “pulling” samples for microbial and endotoxin testing. (adapted from https://www.intechopen.com/chapters/84864)
Sterility and endotoxin testing are mandatory release requirements for drug products (DP) and many drug substances (DS) under United States Pharmacopeia (USP) and U.S. Food and Drug Administration (FDA) guidance. Sterility testing verifies the absence of viable microorganisms, while endotoxin testing ensures pyrogenic lipopolysaccharides from Gram-negative bacteria remain below defined safety limits.
Microbiology groups and cell and gene therapy developers must understand that these tests are not procedural formalities. They directly determine release timing, regulatory compliance, and patient safety.
This article explains:
-
What sterility and endotoxin testing evaluate
-
Where regulatory limits apply
-
Why pre-sterilization bioburden matters
-
When rapid microbiological methods are justified
- How validation and matrix interference affect reliability
This article focuses on sterile biotherapeutics, monoclonal antibodies, and cell and gene therapy products manufactured under aseptic processing.
Why Sterility Is a Critical Quality Attribute
Sterility is a Critical Quality Attribute (CQA) for sterile drug products because viable microorganisms in injectable or implantable therapies can cause septicemia, organ failure, or death.
Under USP, sterility is defined as the absence of viable contaminating microorganisms in a finished product. This requirement applies to parenteral products, ophthalmics, and other sterile dosage forms.
For cell and gene therapies manufactured aseptically, sterility assurance is even more operationally sensitive because terminal sterilization is not feasible.
In practice, sterility testing:
-
Confirms aseptic process control
-
Detects microbial contamination introduced during manufacturing
-
Supports batch release decisions
-
Demonstrates compliance with Good Manufacturing Practice (GMP)
Sterility testing is a regulatory gate for product release and a direct patient safety control. Failure to meet sterility acceptance criteria, results in batch rejection unless a scientifically justified investigation demonstrates laboratory error rather than true contamination.
Sterility assurance is not confined to a single release test performed at the end of manufacturing. In practice, sterility and endotoxin evaluations occur at multiple predefined checkpoints across the entire bioprocess. In a monoclonal antibody workflow, samples are typically pulled during the upstream seed train, after bioreactor harvest, following purification steps, and immediately prior to sterile filtration and bulk fill (Figure 1). These in-process assessments provide real-time control over microbial risk and process hygiene. However, each checkpoint introduces a potential operational bottleneck if sterility and endotoxin methods have not been fully validated, demonstrated for matrix suitability, and operationally transferred to Quality Control in advance.
Regulatory Framework and Microbial Acceptance Criteria
Microbial limits and specified organism requirements are defined in USP and for non-sterile products and sterile products.
The table below summarizes acceptance criteria for specified microorganisms in non-sterile dosage forms under USP:
| Route of Administration |
TAMC (CFU/g or mL) |
TYMC (CFU/g or mL) |
Specified Organisms |
|---|---|---|---|
| Non-aqueous oral | 10³ | 10² | Absence of E. coli (1 g or 1 mL) |
| Aqueous oral | 10² | 10¹ | Absence of E. coli (1 g or 1 mL) |
| Rectal | 10³ | 10² | — |
| Cutaneous / Nasal / Gingival / Auricular | 10² | 10¹ | Absence of S. aureus and P. aeruginosa |
| Vaginal | 10² | 10¹ | Absence of S. aureus, P. aeruginosa, C. albicans |
| Inhalation | 10² | 10¹ | Absence of S. aureus, P. aeruginosa, bile-tolerant Gram-negative bacteria |
TAMC: Total Aerobic Microbial Count
TYMC: Total Yeast and Mold Count
Sterile drug products must meet complete sterility per USP rather than numerical limits.
Pre-Sterilization Bioburden: Why It Matters
Bioburden testing quantifies viable microorganisms present before sterilization. Although pre-sterilization bioburden results are not release criteria, they are used to:
- Evaluate process hygiene
-
Monitor environmental control
-
Confirm sterilization cycle robustness
-
Detect manufacturing drift
Under GMP expectations, trending bioburden supports state of control documentation. A rising bioburden trend may indicate upstream contamination risk even if final sterility passes.
Traditional Compendial Sterility Methods
The USP describes two primary compendial methods:
1. Membrane filtration, preferred for aqueous solutions above 2 mL
2. Direct inoculation, typically used for smaller volumes or products unsuitable for filtration
Testing requires incubation in:
-
Fluid thioglycollate medium (anaerobic bacteria)
-
Soybean-casein digest medium (aerobic bacteria and fungi)
Incubation duration: 14 days minimum.
Positive controls at ≤100 CFU must demonstrate growth promotion. Failure of growth promotion invalidates the test. For aseptically processed products, FDA guidance may extend observational windows or require enhanced justification before release, particularly for cell-based therapies.
Consequently, a sterility failure or invalid test can delay release by at least 14 additional days. For autologous cell therapies, such delays may directly affect clinical scheduling.
Rapid and Alternative Microbiological Methods
The USP permits validation of alternative microbiological methods when equivalence or superiority to compendial methods is demonstrated
FDA supports scientifically validated rapid methods to shorten time to result, especially for advanced therapies.
Examples include:
-
Automated culture detection systems such as BacT/ALERT or BACTEC
-
Nucleic acid amplification methods such as quantitative PCR platforms
-
ATP bioluminescence-based detection
Validation requires:
-
Limit of detection equivalence
-
Specificity against challenge organisms
-
Robustness and reproducibility
-
Matrix interference assessment
Rapid methods reduce release timelines only if fully validated against USP performance criteria. They does not eliminate the need for sterility assurance. It must demonstrate equal or better sensitivity compared to the 14-day method.
Endotoxin Testing Is Independent of Sterility
A sterile product may still contain endotoxins. Therefore, endotoxin testing is mandatory for injectable and implantable products.
Endotoxins are lipopolysaccharides derived from Gram-negative bacterial cell walls. Even if sterilization eliminates viable bacteria, endotoxins may remain because they are heat-stable.
The USP defines:
-
Calculation of endotoxin limits based on dose and route
-
Methods using Limulus Amebocyte Lysate (LAL)
-
Gel-clot, kinetic turbidimetric, and chromogenic assays
Endotoxin exposure can cause fever, hypotension, septic shock, or death. Unfortunately, sterility compliance alone does not ensure pyrogen safety. That is why the FDA Guidance for Industry on Pyrogen and Endotoxins Testing requires demonstration that endotoxin levels remain below established limits before release.
Validation Watch-Outs: Matrix Interference and Method Suitability
Method suitability testing is required under USP 71 and 85 to demonstrate that the product matrix does not inhibit microbial growth or suppress endotoxin detection.
Common challenges include:
-
Antimicrobial excipients inhibiting growth
-
High protein concentrations masking signal
-
Nanoparticle or lipid interference in chromogenic assays
-
PCR inhibitors in rapid sterility platforms
Failure to evaluate interference during development increases the risk of invalid or false-negative release tests. The best practice is to perform recovery and inhibition testing during analytical development and prior to QC transfer.
Conclusion
Sterility and endotoxin testing form the final microbial safety barrier before product release. Under USP and FDA frameworks, sterility confirms absence of viable organisms, while endotoxin testing ensures pyrogen control.
For biologics, monoclonal antibodies, and advanced therapies, method validation, interference testing, and appropriate adoption of rapid microbiological methods directly influence release timelines and regulatory confidence.
Early integration of microbiological method development into CMC strategy reduces downstream release risk and improves operational predictability.
Reviewed by CBS scientific marketing team. March 2026.
Frequently Asked Questions
1. Does a passed sterility test guarantee product safety?
No. Sterility confirms absence of viable microorganisms but does not detect endotoxins or viral contaminants.
2. Can rapid sterility tests replace USP <71>?
Yes, if validated according to USP 1223 and shown to be equivalent or superior in sensitivity and specificity.
3. Is bioburden testing required for sterile products?
Pre-sterilization bioburden is not a release test but is expected for process monitoring and GMP control.
4. Why is the sterility incubation 14 days?
The 14-day period ensures detection of slow-growing or fastidious microorganisms.
5. Are endotoxin limits universal?
No. Endotoxin limits are calculated based on maximum human dose and route of administration under USP 85.
6. Do cell therapies follow the same sterility rules as monoclonal antibodies?
They follow USP 71, but terminal sterilization is not possible, increasing reliance on aseptic process control and rapid testing strategies.
7. What invalidates a sterility test?
Failure of growth promotion controls or laboratory contamination invalidates the run and requires investigation.
